Lilien Lab
Department of Computer Science
Centre for Cellular and Biomolecular Research
University of Toronto
CryoBayes
Navdeep Jaitly, Marcus Brubaker, John Rubinstein, Ryan Lilien
Our method performs ab initio inference of the three-dimensional structures of macromolecules from single particle electron cryo-microscopy experiments using class average images. The main aspects of our approach are as follows:
- We model electron density in real space using tri-linear interpolation over a grid of points.
- Our model is projected on to a set of rotations and the model projections are compared to class averages
- The set of rotations are chosen based on our prior belief about how the macromolecule under study orients itself in the imaging plane.
- A Bayesian framework is used to assess the probability of observing the class averages from a given model
- We optimize our model to find the model that has the highest probability of generating the class averages
Sample Reconstructions
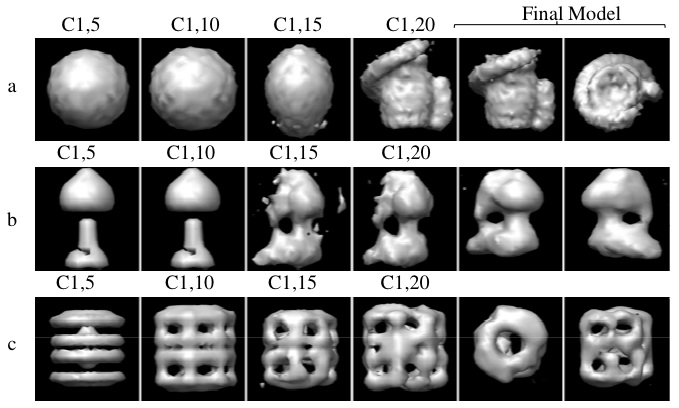
Figure: An illustration of the algorithm's progress towards its 'final' structural model across multiple cycles and iterations for three test systems: (a) A phantom synthetic data set, (b) experimental class averages for ATP synthase, and (c) experimental class averages for GroEL. Cx,y indicates cycle x, iteration y. The columns 'Final Model' show two views of the model returned by our algorithm. This model should serve as a starting point for traditional refinement.
Author Summary: Electron Cryo-Microscopy (Cryo-EM) is an experimental technique for determining the high resolution structures of proteins and protein complexes. The experimental data in a Cryo-EM experiment is a set of extremely noisy projections of the complex along unknown orientations. State of the art methods for structural inference typically consist of two phases, the creation of an initial structural model and then the refinement of this model into a final structure. Here, we present a statistical method to create an initial structural model using a small number of ensemble images. Each ensemble image (or class average) is the average of multiple projections each hypothesized to be taken from the same orientation. Our technique uses a Bayesian or probabilistic approach to infer the molecular structure which has the highest probability of generating the observed images. In other words, our technique identifies the molecular structure that is most consistent with the observed data. Our method is a novel automated solution to a critical step in the Cryo-EM structure determination pipeline. By automatically generating an initial structural model, our software can expedite the structure determination process. To facilitate its use by experimentalists, we have made our software is freely available to the structural biology community.
Spring 2010
CryoBayes beta has been released